GIPR signaling modulates PYY-induced hypophagia and malaise in rodents
The induction of nausea and emesis represents a significant barriers to optimizing weight loss medications for the treatment of obesity. Identifying mechanisms that improve tolerability and/or enhance efficacy without induction of emetic neurocircuitry could provide substantial therapeutic benefits. Candidate peptide YY (PYY)-based approaches for obesity treatment are no exception, as PYY-based therapeutics are uniformly associated with nausea and emesis. Recently, interest in glucose-dependent insulinotropic polypeptide receptor (GIPR)-based therapeutics has resurfaced, with some paradoxical findings from several preclinical studies showing that both GIPR agonism and antagonism, when combined with glucagon-like peptide-1 receptor (GLP-1R) agonists, result in greater body weight loss and superior glycemic control compared to GLP-1R agonism alone. Here, we investigated the effects of pharmacological modulation of the GIPR system on the actions of PYY. We found that systemic GIPR agonism attenuated PYY-induced malaise while preserving its anorectic and body weight-lowering effects in rats. Interestingly, GIPR antagonism enhanced PYY-induced hypophagia and body weight loss without compromising its malaise tolerability profile. Furthermore, inhibition of GIPR signaling significantly reduced PYY-induced c-Fos expression in the area postrema (AP) of the hindbrain. Since both NPY2R and GIPR are expressed in the same AP neurons, this suggests a potential neuronal pathway by which GIPR modulates the effects of PYY. Overall, our findings underscore the multifaceted actions of the GIPR system and highlight the therapeutic potential of both GIPR agonism and antagonism in enhancing and improving the effects of PYY-based obesity treatments.
GIPR agonism lowers nausea, without affecting the hypophagic effects of LA-PYY in rats.
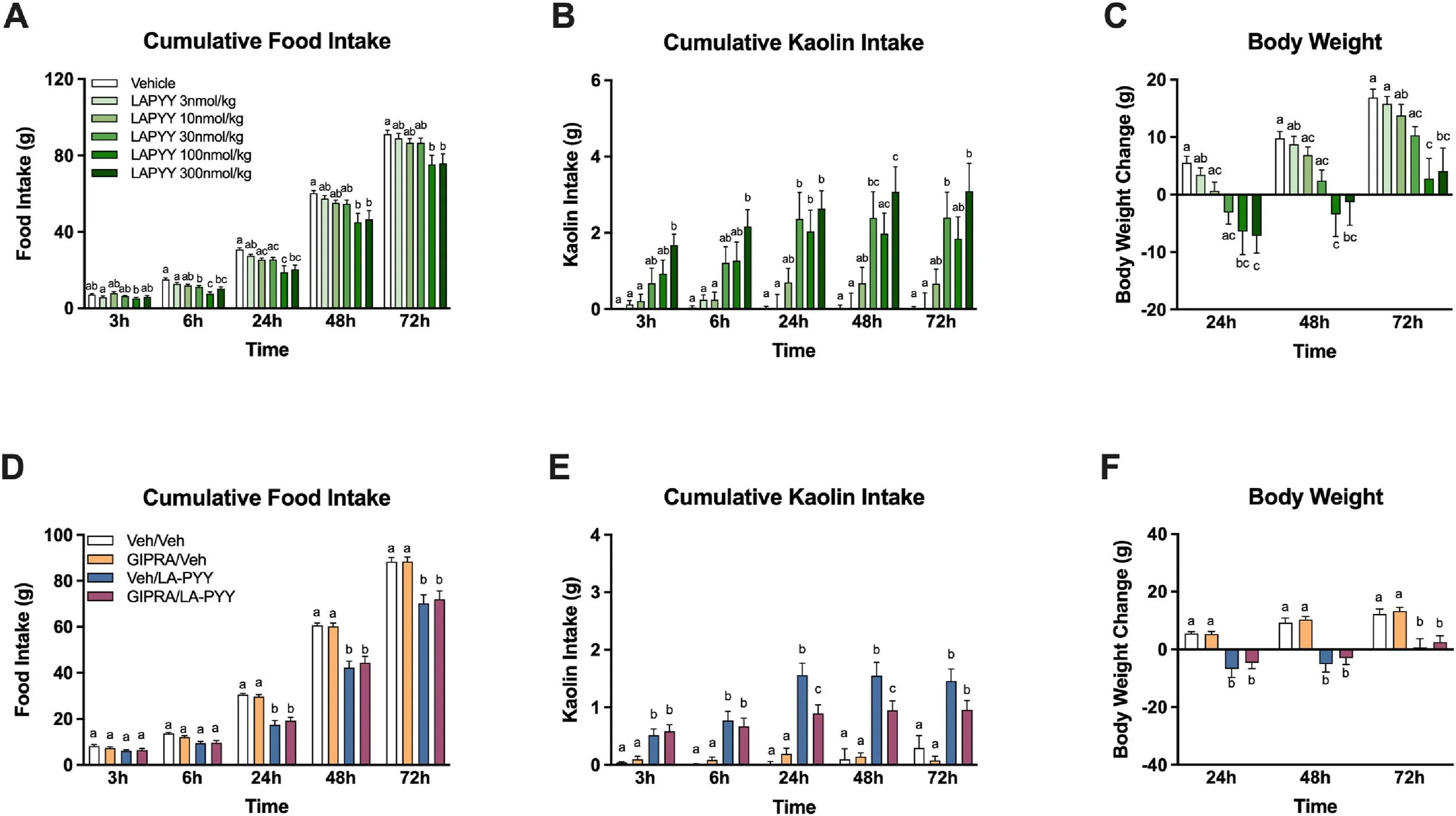
Effects of systemic delivered LA-PYY (3, 10, 30, 100 and 300 nmol/kg, IP, n = 9/group) or vehicle on cumulative food intake (A), daily food intake (B) and kaolin intake (B) and body weight (C). Acute treatment with the GIPR agonist GIP-532 (10 nmol/kg, IP) in combination with LA-PYY (100 nmol/kg, IP) does not modify the food intake-suppressive effects (D) or body weight change (F) as seen with LA-PYY treatment alone. However, pre-treatment does attenuate the LA-PYY-induced kaolin intake, with a significant decrease at 24 h and 48 h (E). n = 12–14/group, data are expressed as means ± SEM. Means with different letters are significantly different from each other (P < 0.05) by two-way repeated measures ANOVA with Tukey's multiple comparison tests.
GIPR antagonism has no acute effects on feeding behaviors and energy homeostasis when administered alone but enhances LA-PYY-induced anorexia and weight loss without aggravating nausea in rats.
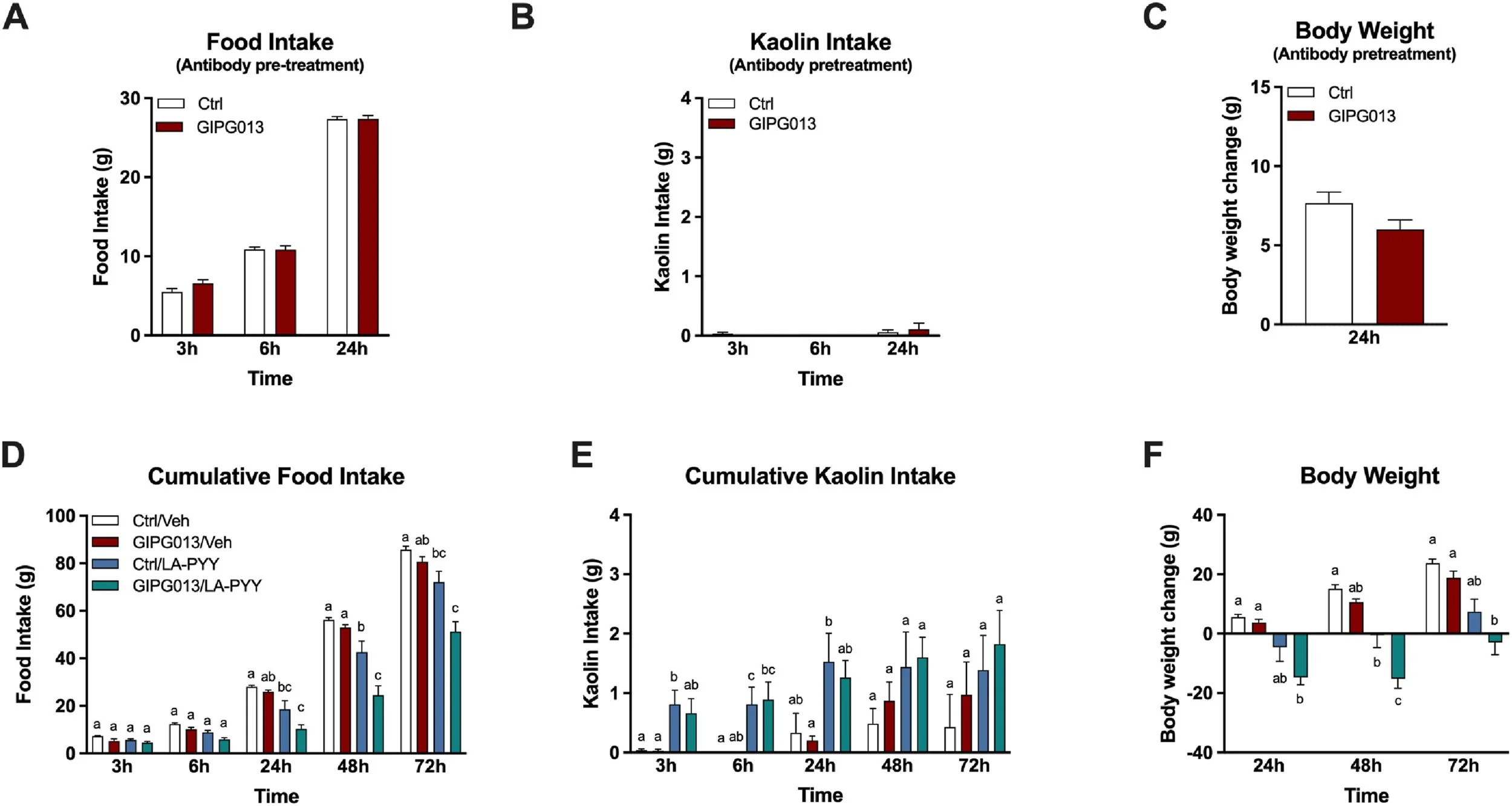
A single injection of GIPR antibody GIPG013 (30 mg/kg, IP) does not affect food intake (A), kaolin intake (B), or body weight (C) (n = 12/group). Pre-treatment of the GIPR antibody GIPG013 (30 mg/kg) increases the anorexia from acute LA-PYY (100 nmol/kg) treatment alone, decreasing food intake (C)and reducing body weight (F). However, pre-treatment does not impact the kaolin intake as seen from acute treatment of LA-PYY alone (E) (n = 7–8/group). Data are expressed as means ± SEM. Data in (A and B), analyzed with two-way repeated measures ANOVA, followed by Sidak's multiple comparison tests, data in (D, E, and F) analyzed with two-way repeated measures ANOVA, followed by Tukey's multiple comparison tests, data in (C) tested with an unpaired t test. Means with different letters are significantly different from each other (P < 0.05).